Humanization of Yeast Genes (Part 2)
- 5 minsAbout a month ago I wrote a post in which I mentioned I would be working on a project to try to determine if pathogenic mutations in genes shown on a paper by the Marcotte lab to be replaceable by the human version of the gene, which means that the yeast is able to survive, are in amino acids that are more conserved than the average amino acid in the protein.
I was initially interested in monogenic diseases, so I parsed through the GWAS catalog and the OMIM genemap to try to find monogenic diseases. The OMIM genemap contained 1332 conditions/symptoms that are associated with a single gene. The GWAS catalog contained 83. I compared these lists against the lists of replaceable and non replaceable genes and got no matches.
I then looked at a list containing 260 genes associated with mendelian diseases that affect all organelles. I got 8 matches for replaceable genes (ALG1, DDOST, DHDDS, DOLK, DPAGT1, DPM1, PIGA, SPTLC1) and 6 matches for non replaceable genes (ALG2, COG4, INPP5E, MPI, PIGM, RFT1)
I then proceeded to analyze amino acid conservation across organisms (human, mice, zebrafish, drosophila, c. elegans and yeast) as described in my previous post (where I analyzed mutation conservation across organisms for the NPC1 gene)
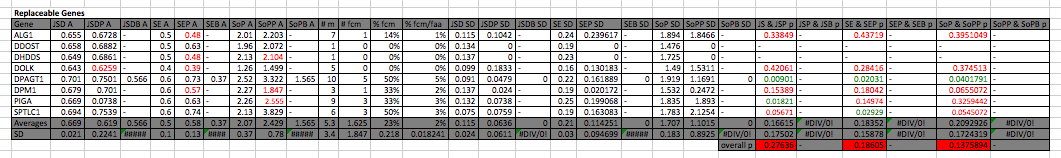
Conservation in replaceable genes
In both Shannon Entropy and Sum of Pairs pathogenic mutations are (on average) in amino acids that are more conserved than the average amino acids in the protein. The difference between pathogenic and average amino acid conservation isn’t statistically significant when looking at all the genes (Jensen Shannon Divergence p: 0.2763591, Shannon Entropy p: 0.1860548, Sum of Pairs p: 0.1375894), though it is in DPAGT1 throughout all 3 methods (Jensen Shannon Divergence p: 0.009011903, Shannon Entropy p: 0.02030809, Sum of Pairs p: 0.04017908), in PIGA using Jensen Shannon Divergence (p: 0.01821001) and on SPTLC1 using Shannon entropy (p: 0.02929091; in all of these pathogenic mutations are more conserved) . The average number of mutations per gene is 5.25, there was only one benign mutation analyzed (so we can’t really make any conclusions about benign mutations). On average 1.625 mutations are fully conserved (23%) and the fully conserved mutations make up on average 2% of the fully conserved amino acids.
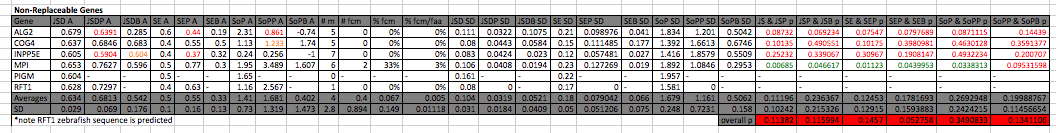
Conservation in non-replaceable genes
Throughout all 3 methods pathogenic mutations are on average more conserved than the average amino acid in the protein, though the difference is not statistically significant (Jensen Shannon Divergence p: 0.113819, Shannon Entropy p: 0.145702, Sum of Pairs p: 0.3490833) when looking at all the genes. Pathogenic mutations are also in amino acids that are on average more conserved than amino acids containing benign mutations, though the difference is again not statistically significant (Jensen Shannon Divergence p: 0.1159944, Shannon Entropy p: 0.05275803, Sum of Pairs p: 0.1341106) In MPI the difference between the average amino acid and the amino acids containing pathogenic mutations is statistically significant across all methods (Jensen Shannon Divergence p: 0.006847529, Shannon Entropy p: 0.01122596, Sum of Pairs p: 0.03383133), and the difference between pathogenic and benign mutations is statistically significant using Jensen Shannon Divergence and Shannon Entropy (p: 0.04661734 and 0.04399531 respectively) but not Sum of pairs (p: 0.09531598). The average number of mutations per gene is 4, with on average 24% being benign. On average 0.4 mutations are fully conserved (7%) and the fully conserved mutations make up on average 1% of the fully conserved amino acids.
Relationship between replaceable and non replaceable genes
As expected, on average amino acids in replaceable genes are more conserved than amino acids in non replaceable genes. The difference is statistically significant across all methods (Jensen Shannon Divergence p: 0.0289762, Shannon Entropy p: 0.03484218, and Sum of Pairs p: 0.04859736). On average pathogenic mutations are more conserved in replaceable genes than non replaceable genes throughout 2 out of 3 methods (Shannon Entropy and Sum of Pairs) though the difference is not statistically significant (p: 0.3611836 and 0.1574445 respectively). Benign mutations are also more conserved in replaceable genes than in non replaceable genes but I did not have a large enough sample size (n = 1) on replaceable genes to determine statistical significance.

Conclusion and next steps
It is quite possible that statistical significance was not reached because of the small amount of mutations these genes contained, in addition to the small amount of genes that I looked at. The next step will be to look at all the genes in the list of 260 genes associated with mendelian diseases that affect all organelles. Maybe something else that would be interesting to do is to look at all the genes in the list of replaceable and non replaceable genes in the paper and that might give statistically significant results.
References
Kachroo, AH et al. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 348(6237) 921-925 (2015)
http://edit.tigem.it/en/research/researchers/antonella-de-matteis/disease-genes
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014 Jan 1;42(1)
Thompson, J D, D G Higgins, and T J Gibson. “CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice.” Nucleic Acids Research 22.22 (1994): 4673–4680. Print.
Capra JA and Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics, 23(15):1875-82, 2007

Jamie Cayley
Grad Student & TA at Missouri State University