Mutation Conservation Across Organisms in NPC1
- 5 minsAs I mentioned in my previous post, something really important while using model organisms (I used yeast as an example but this is true for every model organism, like mice, flies, fish, worms) is figuring out whether the results are translatable. Given that I am interning at Perlstein Lab and they are interested in Niemann-Pick Disease, I decided to see if mutations in NPC1 are in conserved amino acids.
I got the protein sequences for NPC1 in humans, mice, drosophila, c. elegans, zebrafish, and yeast from Entrez Protein. I then performed a multiple sequence alignment (MSA) using CLUSTALW2. The way MSA typically works is that you have a set S of k sequences
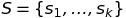
Let M be an MSA for the set S of k sequences. The alignment of s_i with s_j induced by M is generated as follows:
Remove from M all rows except i and j, then remove all columns that contain only blanks
The sum of pairs score of M is the sum of all pairwise induced alignment scores. The optimal MSA for S with respect to sum of pairs is the MSA with the highest sum of pairs score out of all possible MSA for S. Then to compute the best possible alignment d(i,j) for every pair of prefixes (lengths i, j) we use the following formula

The problem with this method is that it quickly gets complex; to compute the best possible alignment d(i,j,k) for every triple of prefixes (lengths i,j,k) we would have to use the following formula

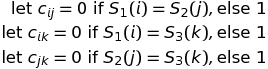
For k sequences of length n we have to perform the following number of computations
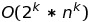
CLUSTALW2 uses a two step method that is faster. The main idea is to find strong signals (highly conserved blocks) first, outliers are added last and it increases the chances that conserved blocks survive.
The first step is to compute a guide phylogenic tree. To do this it computes all pairwise alignments and stores them in a similarity matrix M
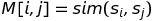
It then computes the guide tree using hierarchical clustering. It chooses the smallest M[i,j] then forms a new branch of the tree with s_i and s_j. It then computes the distance from the ancestor of s_i and s_j to all other sequences as the average of the distances to s_i and s_j; to do this it sets M’ = M, deletes rows and columns i and j, adds a new column and row (ij) and then 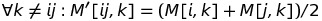
It finally iterates until M’ has only one column/row
The second step is progressive MSA. It computes pairwise alignment in the order given by the guide tree. To align a MSA M_1 with a MSA M_2 it uses the usual global alignment algorithm and to score a column it computes the average score over all pairs of symbols in the column.
After all this we end up having to perform less computations
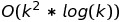
You can see a part of the alignment file below.

I got a list of mutations in the NPC1 gene from ClinVar. I got a total of 51 amino acid changes, 6 of which were benign/likely benign. I then searched for the amino acid in which the mutations occur in the sequence alignment and saw whether the amino acid was conserved or not.
To see if the amino acids were conserved I used 3 different methods: Jensen Shannon Distribution, Shannon Entropy and Sum of Pairs. For consistency Shannon entropy scores were scaled to the range [0,1] and then subtracted from 1, so that higher scores indicate greater conservation.
To see if pathogenic mutations are in amino acids that are more conserved than average and more conserved than benign mutations, I calculated the average scores across the entire protein (1278 amino acids for the human protein), the amino acids implicated in pathogenic mutations, and the amino acids implicated in benign mutations.

As expected the average score for pathogenic mutations was higher than the average for the entire protein and the benign mutations. To figure out if the difference was statistically significant I then calculated the standard deviation for these values and the p values for the relationships whole protein - pathogenic and pathogenic - benign and found that the difference between all of these relationships throughout the 3 methods for pathogenic - benign and 2 methods (Jensen-Shannon Divergence, Shannon Entropy) for whole protein - pathogenic were statistically significant (p < 0.05)
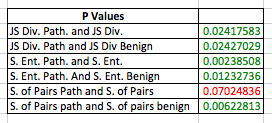
You can see the full mutation data here: https://github.com/materechm/Bioinformatics/blob/gh-pages/NPC1MutationConservationData.xlsx
The commands I used to generate the p values on R here: https://gist.github.com/materechm/a673f4034f7a087ae04a
And the alignment file here: https://github.com/materechm/Bioinformatics/blob/gh-pages/proteinAligmentNPC1.pdf
If you have any comments or suggestions on interesting things that can be done with this data shoot me an email at maria@perlsteinlab.com
References
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014 Jan 1;42(1)
Thompson, J D, D G Higgins, and T J Gibson. “CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice.” Nucleic Acids Research 22.22 (1994): 4673–4680. Print.
Capra JA and Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics, 23(15):1875-82, 2007

Jamie Cayley
Grad Student & TA at Missouri State University