Mutation Conservation in Mendelian Genes
- 5 minsThis is a follow up to the mutation conservation analyses I’ve been doing (you can see the analysis for NPC1 here and for a set of genes that were found to be replaceable here)
For this analysis I started off with a list of 260 genes associated with mendelian diseases that affect all organelles (the list came mainly from here). Then, for each gene I downloaded the protein sequences for human, mouse, drosophila, zebrafish, c. elegans and yeast (as many as were available) from HomoloGene. If a sequence was missing I checked on OMA browser as well.
Afterwards I created a multiple sequence alignment using CLUSTAL W2. I then used Princeton’s Protein Residue Conservation Prediction tool to calculate the Jensen Shannon Divergence, Shannon Entropy and Sum of Pairs scores throughout the protein. I proceeded to search Clinvar for mutations associated with the gene of interest and inputed this information to a program I wrote.
Results: first 130 genes
Throughout all methods pathogenic mutations are on average more conserved than the average amino acid in the protein (Jensen Shannon Divergence p: 3.19E0-5, Shannon Entropy p: 6.06E-09, Sum of Pairs p: 2.77E-09), and more conserved than benign mutations (Jensen Shannon Divergence p: 2.75E-10, Shannon Entropy p: 4.51E-18, Sum of Pairs p: 6.32E-18)
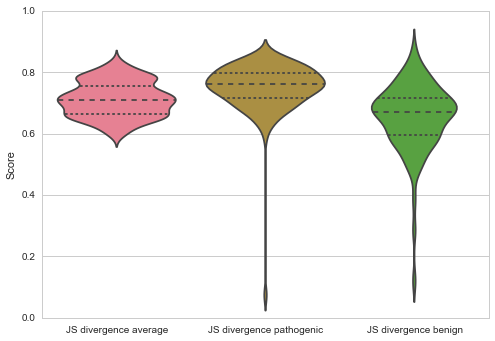
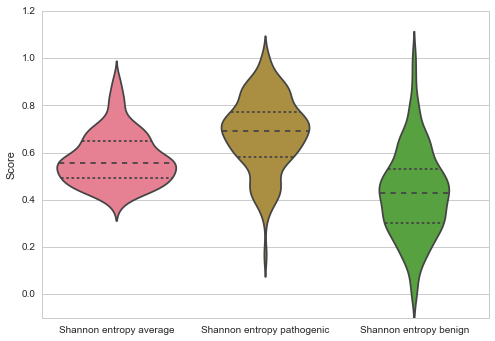
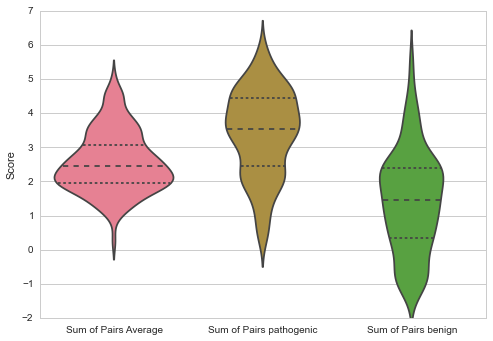
The average number of mutations per gene is 12.10852713, and 21% are benign. On average 5.611111111 mutations are fully conserved which make up about 3% of the fully conserved amino acids on the proteins.
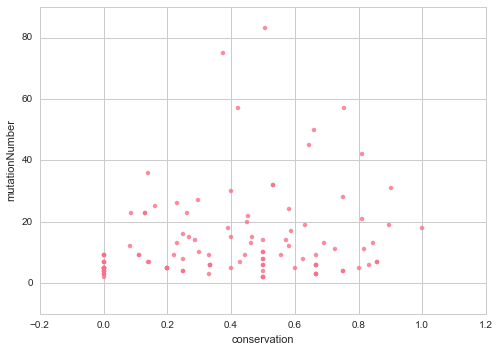
Notably, the difference in conservation between pathogenic mutations and the average amino acid seems to be more significant when we have more mutations, and the relationship (pearson correlation: -0.397501) appears to follow a power trend, which means we get a stronger signal as mutations increase as expected, which again could be the reason why we did not reach statistical significance on our (previous mutation conservation analysis)[http://mtc.science/humanization-of-yeast-genes-part-2]
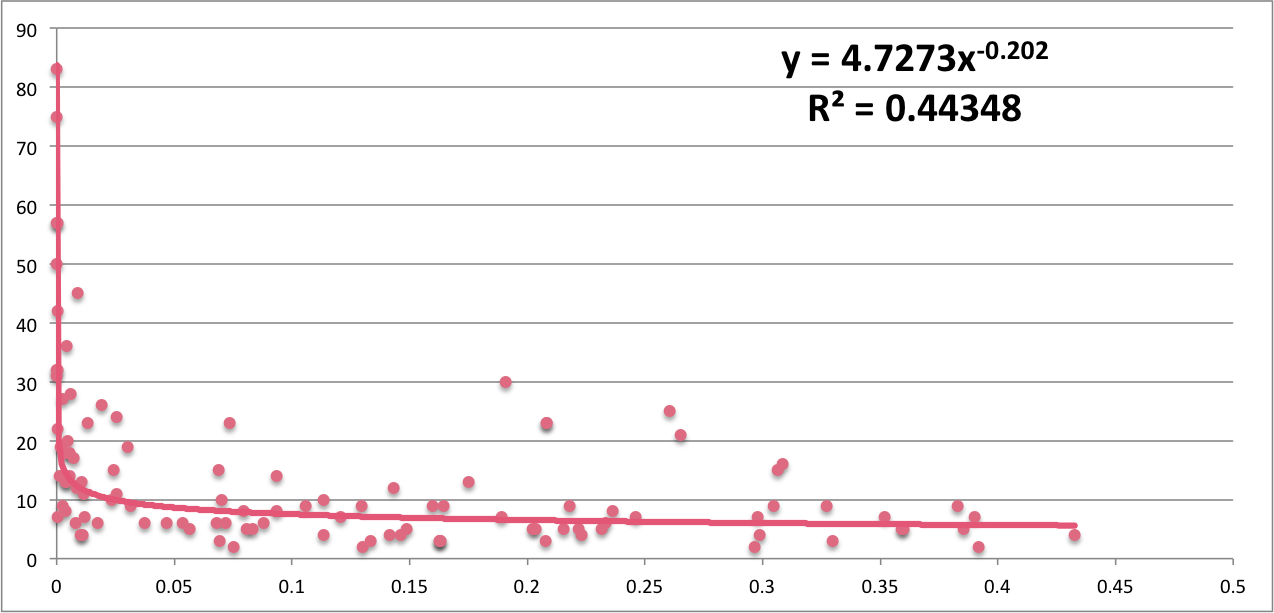
There does not seem to be a relationship between number of mutations and percentage fully conserved mutations. This means that even though the difference in conservation between pathogenic and the average is more significant as mutation number increases, the percentage of fully conserved mutations does not depend on how many mutations we have.
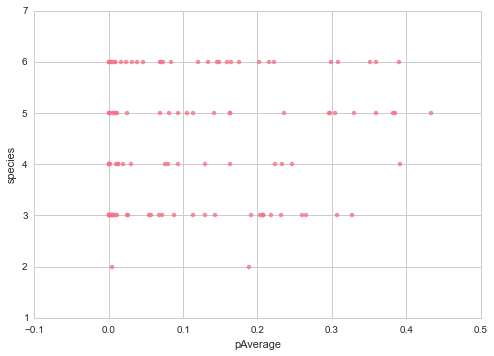
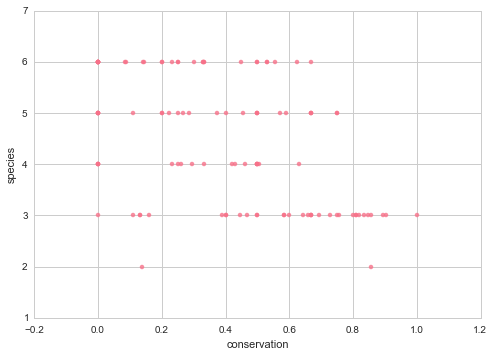
And there does not seem to be a correlation between the percentage fully conserved mutations or the p value with the number of species where there was a homolog, which means we aren’t getting a stronger signal just because there are less species.
Some visual examples of mutation conservation
In the following graphs amino acids highlighted in purple indicate fully conserved pathogenic mutations, in red are mutations that are not fully conserved and all amino acids with an * below are fully conserved.
1) CLCN5
CLCN5 has 18 pathogenic mutations, all of which are fully conserved and make up 2.80% of the fully conserved amino acids in the protein. Pathogenic mutations are more conserved than the average amino acid in the protein (Jensen Shannon Divergence p: 0.013477584, Shannon Entropy p: 3.35166E-09, Sum of Pairs p: 0.002770766)

2) CLN3
CLN3 has 29 pathogenic mutations, 58.62% which are fully conserved and make up 21.79% of the fully conserved amino acids in the protein. Pathogenic mutations are more conserved than the average amino acid in the protein (Jensen Shannon Divergence p: 0.000347195, Shannon Entropy p: 0.000670302, Sum of Pairs p: 0.000207668)
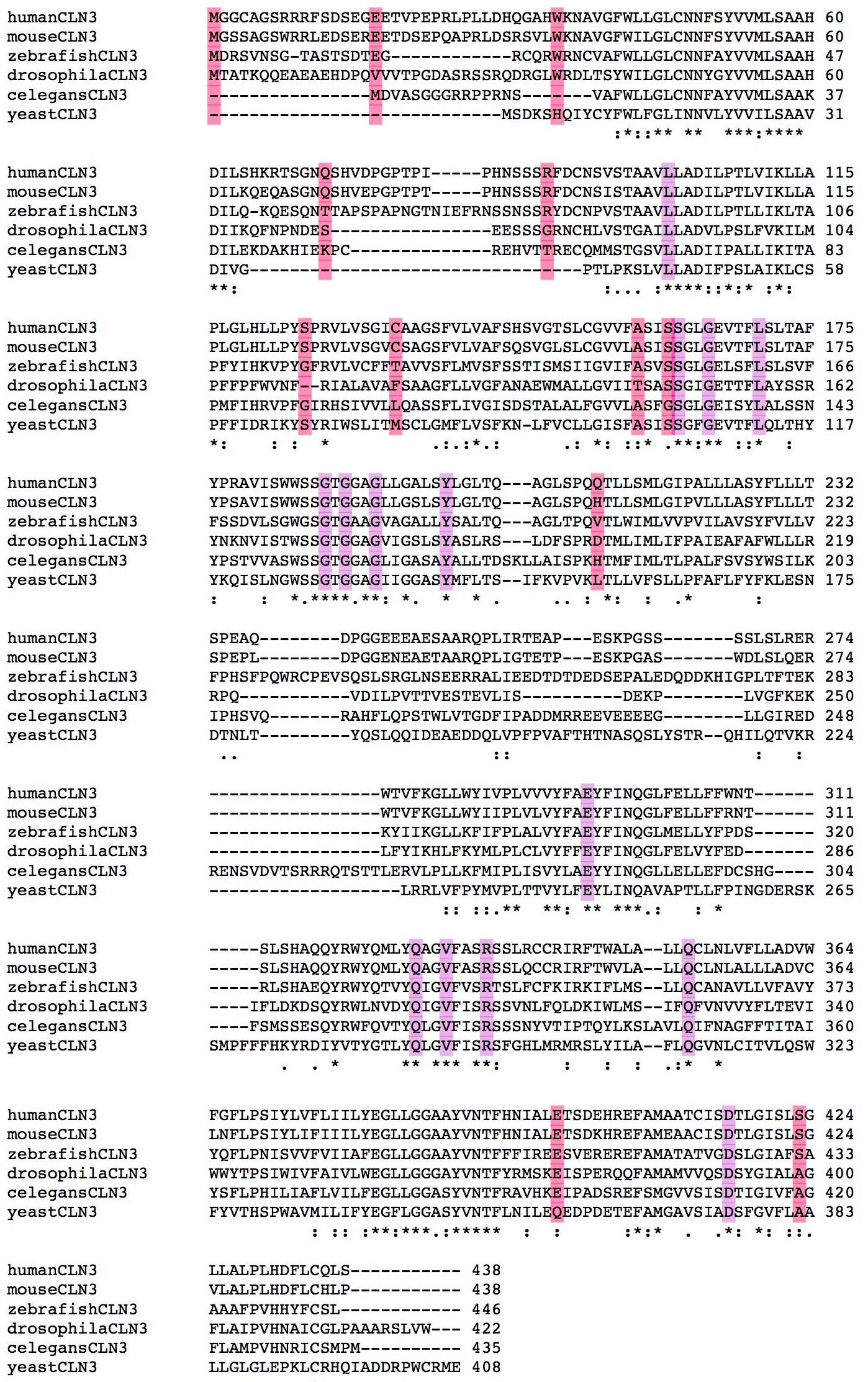
3) DHCR7
DHCR7 has 30 pathogenic mutations, 93.33% which are fully conserved and make up 8.16% of the fully conserved amino acids in the protein. Pathogenic mutations are more conserved than the average amino acid in the protein (Jensen Shannon Divergence p: 1.15794E-05, Shannon Entropy p: 1.36185E-05, Sum of Pairs p: 0.000107308)
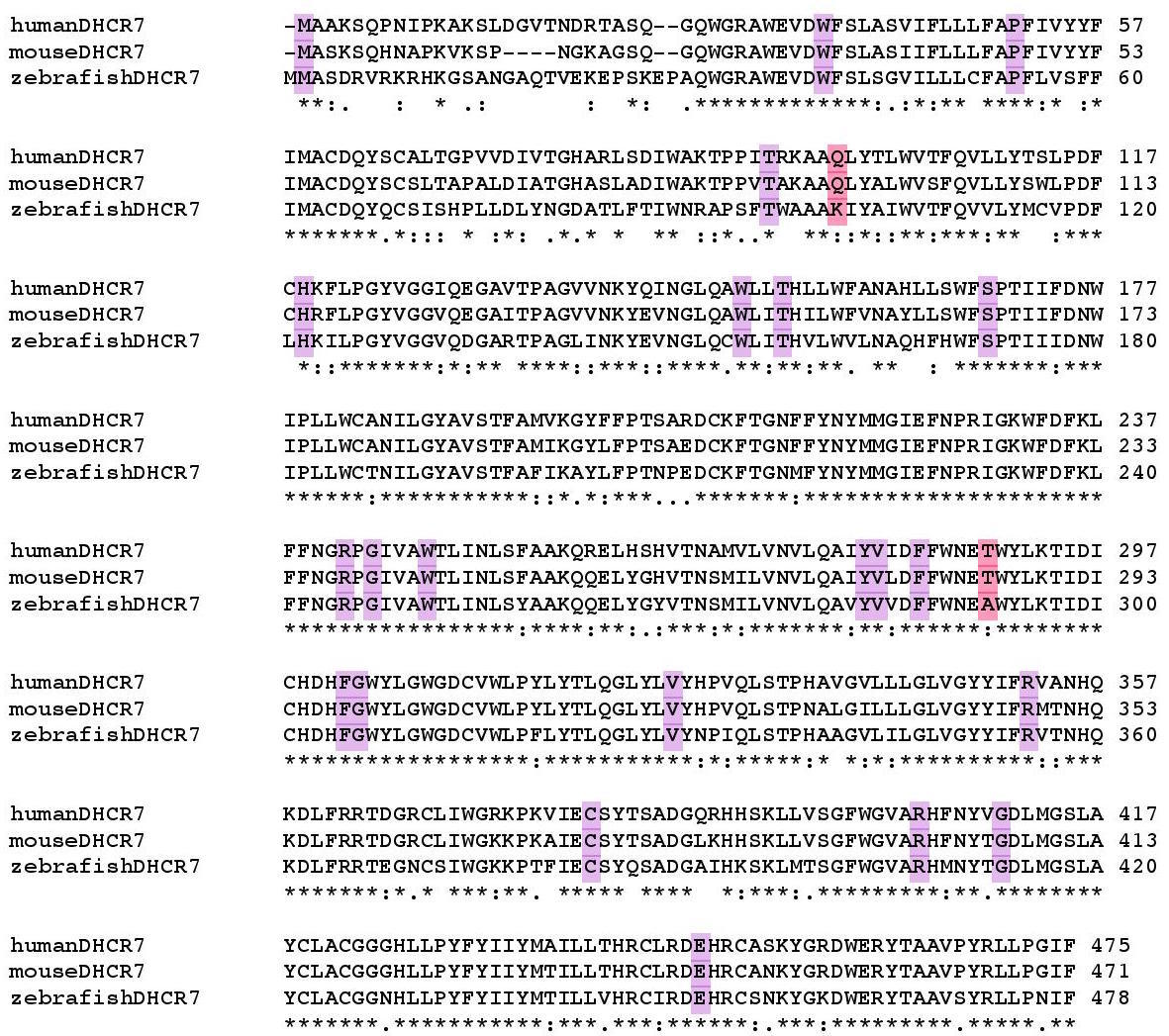
4) DNM2
DNM2 has 17 pathogenic mutations, all of which are fully conserved and make up 2.39% of the fully conserved amino acids in the protein. Pathogenic mutations are more conserved than the average amino acid in the protein (Jensen Shannon Divergence p: 0.003927946, Shannon Entropy p: 6.44148E-08, Sum of Pairs p: 0.000504219)

5) HEXA
HEXA has 40 pathogenic mutations, 85.00% are fully conserved and make up 12.45% of the fully conserved amino acids in the protein. Pathogenic mutations are more conserved than the average amino acid in the protein (Jensen Shannon Divergence p: 0.001007446, Shannon Entropy p: 3.23795E-06, Sum of Pairs p: 1.93499E-06)
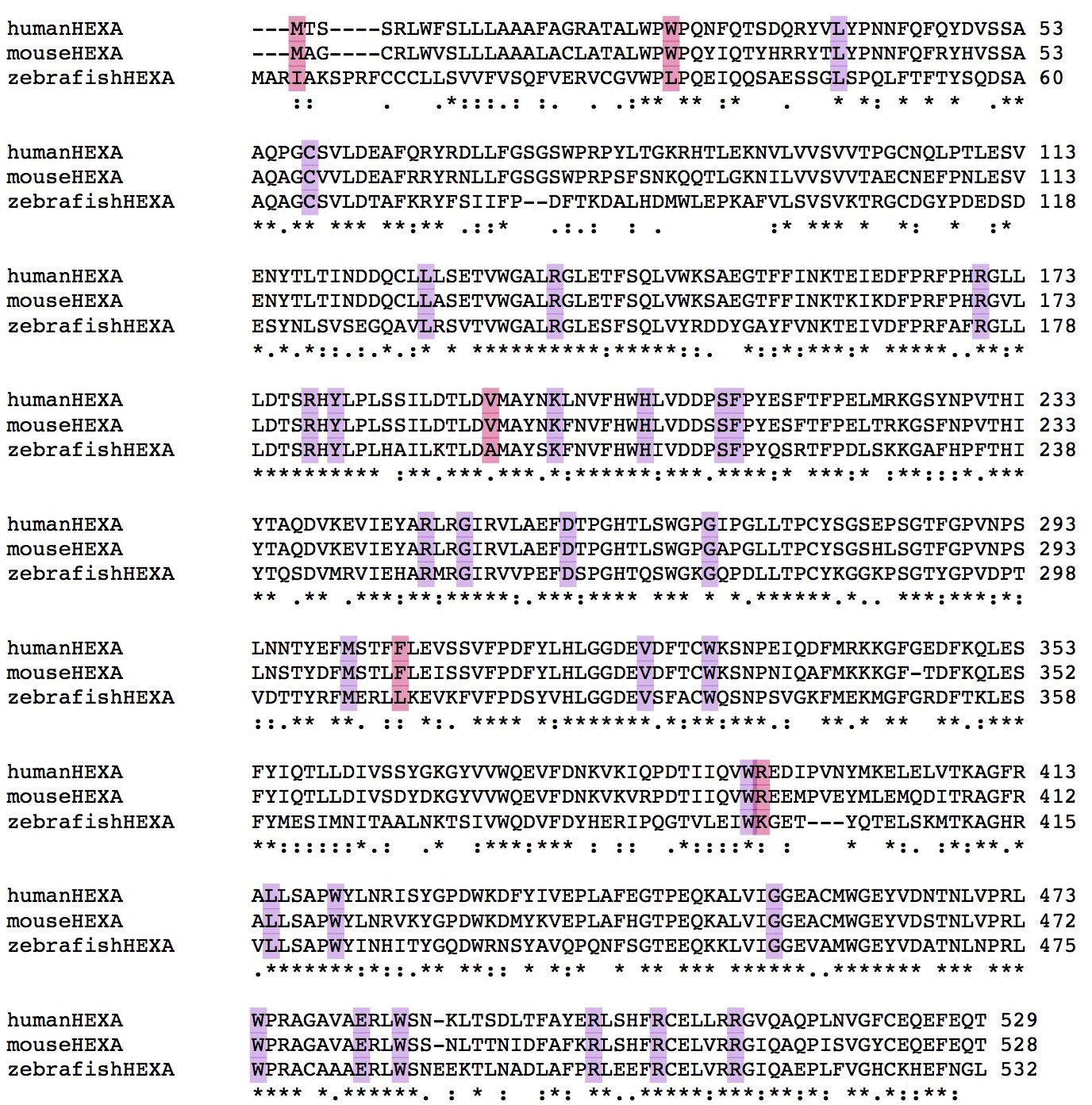
Data
All data is here: https://github.com/materechm/plabData
References
http://edit.tigem.it/en/research/researchers/antonella-de-matteis/disease-genes
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014 Jan 1;42(1)
Thompson, J D, D G Higgins, and T J Gibson. “CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice.” Nucleic Acids Research 22.22 (1994): 4673–4680. Print.
Capra JA and Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics, 23(15):1875-82, 2007

Jamie Cayley
Grad Student & TA at Missouri State University