Playing Around With NGS
- 5 minsHere I will walk you through how to take reads from a sequencer and assemble/map a genome and generate a VCF file + do some functional predictions.
Set-up
Install/download samtools (1.2), bwa, picard (sourceforge version), GATK + Java 7 (won’t work with other versions of Java) and SnpEFF You’ll need a computer that can run UNIX and about 50 GB of hard disk space (+ a fast internet connection makes things way better)
Getting test data
In terminal type "ftp ftp.broadinstitute.org" (username: gsapubftp-anonymous, password: your email); change directory (cd) to /bundle/2.8/hg19 and download ucsc.hg19.fasta.gz with the get command and uncompress the reference with the gunzip command. Then download dbsnp_138.hg19.vcf.gz and it’s idx from the same location.
Repeat the above steps with ftp-trace.ncbi.nih.gov (username: anonymous, password: your email); navigate to /giab/ftp/technical/garvan_data and download all the .fastq files that don’t say “trimmed” e.g. "NIST7035_TAAGGCGA_L001_R1_001.fastq.gz"
Pair end Sequence Assembly
[Note - for running bwa and samtools commands it might be necessary to be on the directory where bwa or samtools is located and use ./bwa or ./samtools]
The first thing we need to do in order to use bwa is construct an index for our reference genome. We do this by running the following command:
bwa index -a bwtsw [reference.fasta]. Once the reference index is built, sequence alignment indices of our fastq files need to be built, we do that with the following commands: bwa aln [reference.fasta] [short_pair_1.fastq] > [short_pair_1.sai] bwa aln [reference.fasta] [short_pair_2.fastq] > [short_pair_2.sai]
We can now combine our reference index, the two alignment indices, and our two fastq files to create a single sequence alignment map
bwa sampe [reference.fasta] [short_pair_1.sai] [short_pair_2.sai] [short_pair_1.fastq] [short_pair_2.fastq] > [short_pair.sam]
Next we need to change “” to “o” as a flag for unaligned reads, ajust mate pair info and add read headers, which can all be done with Picard using the following commands java -Xmx[memory]g -jar ValidateSamFile.jar I=[.bam or .sam] This command should display MAPQ flag error, mate-pair error, and header error java -Xmx[memory]g -jar FixMateInformation.jar I=[.bam] O=[.bam] VALIDATION_STRINGENCY=LENIENT
java -Xmx[memory]g -jar ValidateSamFile.jar I=[fixed.bam] IGNORE=INVALID_MAPPING_QUALITY Next we need to validate file accepting MAPQ flag “” java -Xmx[memory]g -jar AddOrReplaceReadGroups.jar I=[.bam] O=[.bam] LB=anything PL=illumina PU=anything SM=anything
Next we need to re-sort the picard output and we’ll get as an output an unsorted bam file which has to be converted to binary and sorted using samtools. To sort it we use the following commands: samtools view -bS -o [output.bam] [input.bam or .sam] samtools sort [input.bam] [output.sorted.bam]
To view the bam file we use the command samtools tview file.bam (if the file appears all white you can press n to see colors. You should see something like this:

The VCF file
A VCF file (Variant Call Format) is a text file containing information about the mutations in our genome.
The first thing we need to do is generate the VCF file, which on samtools 1.2 is generated using the following command: ./samtools mpileup -ugf [reference.fasta] [sorted.bam] |./bcftools/bcftools call -vmO z -o [output.vcf]
To use GATK we’ll need to generate a .dict and a .fasta.fai file to allow efficient random access to the reference. To generate the fasta.fai we use the following samtools command samtools faidx reference.fa and to generate the .dict we use the following Picard command java -jar CreateSequenceDictionary.jar -REFERENCE=reference.fa -OUTPUT=reference.dict
Next we’ll use the dbsnp file we downloaded at the beginning to annotate the VCF file (and add the rsID for the mutations that are both in our VCF and the dbsnp, to do that we run the following GATK command java -Xmx[memory]g -jar GenomeAnalysisTK.jar -R [ref.fasta] -T VariantAnnotator -o [output.vcf] --variant [input.vcf] --dbsnp [dbsnp.vcf]
Next we’ll run variant filtration to create a couple of filters that tell us how confident we are on the mutation; to do that we run the following GATK command `java -Xmx[memory]g -jar GenomeAnalysisTK.jar -R [reference.fasta] -T VariantFiltration -o [output.vcf] --variant [input.vcf] --filterExpression "DP > 10 && DP < 25" --filterName "low" --filterExpression "DP > 25 && DP < 50" --filterName "mid" --filterExpression "DP > 50 && DP < 150" --filterName "high" --filterExpression "DP > 150" --filterName "Xtreme" --filterExpression "DP < 10" --filterName "PASS"
Troubleshooting
If you can’t run variant annotator because the variant contigs are not the same as the reference contigs run the following (with picard 1.121 or greater) java -Xmx2g -jar picard.jar SortVcf INPUT=[input.vcf] OUTPUT=[output.vcf] SEQUENCE_DICTIONARY=[reference.fasta] Then you might have to edit the output vcf and remove the #contigs from the header.
Functional predictions with SnpEFF/GATK
Finally we’ll use SnpEFF and GATK to generate some functional predictions. We just need two commands to do this: java -Xmx[memory]G -jar snpEff.jar -c snpEff.config -v -o gatk hg19 [input.vcf] > [output.snpeff.vcf] (run with snpEFF) and with GATK java -Xmx[memory]g -jar GenomeAnalysisTK.jar -T VariantAnnotator -R [ref.fasta] -A SnpEff --variant [input.vcf (same input than the last algorithm)] --snpEffFile [input.snpeff.vcf] -L [input.vcf] -o [output.gatk.vcf]
At the end your VCF should look similar to this:

And the SnpEFF output should look similar to this: http://materechm.github.io/Bioinformatics/snpEFF_summary.html
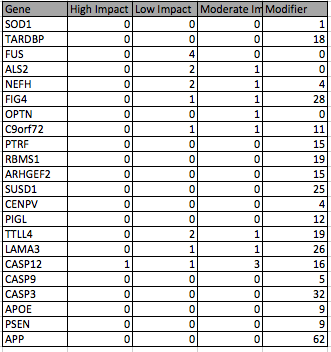
You can get a lot of information from the SnpEFF output, for example I decided to look into ALS and AD mutations and generated a table and a graph with the number of mutations in some genes associated these diseases divided by their functional impact. You can see the table and graph below:

Jamie Cayley
Grad Student & TA at Missouri State University